

Advanced SAR studies to guide and accelerate lead optimization
Using SAR and QSAR, we help you pinpoint key structural features, optimize lead compounds, and accelerate your path toward a viable candidate, backed by a team of experts who challenge assumptions and pursue the most insightful solutions. Apply our deep medicinal chemistry expertise and state-of-the-art computational methods to SAR studies to systematically explore the structure-activity relationships of your molecule.
The role of SAR in the drug discovery critical pathway
Structure-Activity Relationship (SAR) analysis is a fundamental step in drug discovery. The role of SAR studies involves systematically exploring how modifications to a molecule’s structure affect its biological activity and ability to interact with a target of interest. By designing and testing a series of related compounds, researchers can identify key structural features that influence potency, selectivity, and safety, allowing progression from initial hits to well-optimized lead compounds.
SAR studies not only guide lead optimization but also help highlight potential safety issues early, providing valuable data for developing computational models to predict the activity of new compounds. By uncovering these structure-activity relationships, you can accelerate your drug discovery process and maximize your chances of identifying viable, effective drug candidates.
Working with the Oncodesign Services-ZoBio Group
At the Oncodesign Services-ZoBio Group, we offer tailored SAR and QSAR model building services to accelerate your drug discovery process. Our team of experts includes computational and medicinal chemists with decades of experience in drug development.
We combine advanced computational chemistry platforms to accelerate and de-risk drug discovery projects. Our expertise spans SAR and QSAR analysis, powered by the Molecular Operating Environment (MOE), which seamlessly integrates Structure-Based (SBDD) and Ligand-Based Drug Design (LBDD) approaches. Workflow automation through KNIME further enhances efficiency, enabling high-throughput and reproducible in silico screening.
To gain deeper insight into molecular interactions and optimize compound design, we routinely apply molecular dynamics simulations using NAMD. This allows us to explore the dynamic behavior and stability of ligand–protein complexes, providing you with a powerful edge in understanding and fine-tuning molecular mechanisms at atomic resolution.
Key SAR analysis services
Using a combination of experimental and computational methods, our medicinal and computational chemists can support:
- Design and synthesis of SAR series: Developing a systematic set of compounds with targeted structural variations.
- Biological data analysis: Interpreting the results of biological assays in the context of structural changes.
- Activity cliff identification: Pinpointing small structural changes that lead to large activity differences.
- Computational SAR/QSAR modeling: Using software and algorithms to build predictive models that link chemical structure to biological activity.
- Molecular docking and dynamics simulations: Investigating how compounds interact with their biological targets at a molecular level.
- Scaffold hopping and design of novel chemical entities: Generating ideas for new chemical structures with desired properties.
- Integration of early-ADME and selectivity data: Interpreting the results of early-ADME assays and off-target effects to guide optimization.
- Expert consultation and interpretation: Providing specialized knowledge and insights to guide drug discovery projects.
Experimental SAR studies
Experimental SAR studies involve the synthesis and testing of a series of compounds that are structurally related to a known active compound. The biological activity of each compound is measured, and the results are analyzed to identify structural features that are associated with increased or decreased activity. This information can then be used to design new compounds with improved biological properties.
Specific experimental SAR techniques include:
- Biological assays: These assays measure the biological activity of a compound on a target, such as an enzyme, receptor, or cell.
- Pharmacokinetic studies: These studies measure the absorption, distribution, metabolism, and excretion of a compound in the body.
- Toxicological studies: These studies assess the safety of a compound by measuring its effects on various organs and systems in the body.
Computational SAR studies
Computational SAR methods utilize machine learning models to predict the biological activity of new compounds based on their chemical structure. These models are developed using data from experimental SAR studies. The models can then be used to identify promising lead compounds and to optimize their structure for improved activity and selectivity.
Specific computational SAR techniques include:
- Molecular modeling: These techniques use computer software to build and simulate three-dimensional models of molecules. This information can be used to understand how molecules interact with their biological targets.
- Quantitative structure-activity relationship (QSAR) modeling: These models use artificial intelligence models to relate the chemical structure of a compound to its biological activity.
We use a combination of computational chemistry software to evaluate SARs and QSARs. Our Molecular Operating Environment (MOE) software combines Structure-Based Drug Design (SBDD) and Ligand-Based Drug Design (LBDD) approaches efficiently. Our software is further assisted by Konstanz Information Miner (KNIME), which allows automatization and speeds up computational LBDD and SBDD workflows.
SAR analysis methodology
SAR describes the direct relationship between the chemical structure of a molecule and its biological or physicochemical activity. Key aspects of SAR studies include:
-
Core principle
The basic premise of SAR is that the specific arrangement of atoms and functional groups within a molecule dictates its properties and how it interacts with biological systems (e.g., proteins, enzymes, receptors, DNA). Therefore, even small changes to a molecule’s structure can lead to significant alterations in its biological activity, including its potency, selectivity, metabolic stability, and toxicity.
-
SAR study workflow
SAR studies typically involve:
- Synthesis of analogs: A series of compounds (analogs) are synthesized, each with slight structural modifications from a known active compound (a “hit” or “lead” compound).
- Biological testing: The biological activity of each analog is measured (e.g., enzyme inhibition, receptor binding, antimicrobial activity, cellular effect, in vivo efficacy).
- Analysis and correlation: The results are analyzed to identify which structural features are associated with increased, decreased, or altered activity. This helps to pinpoint the “pharmacophore” – the essential features of a molecule responsible for its biological action.
- Structural information and design: Finally, structural information obtained through crystallography, NMR or cryo-EM is used to rationalize observed SAR and design the next set of compounds.
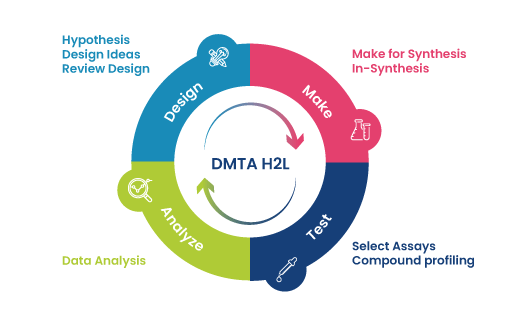
Pictured above: Design – Make – Test – Analyze Cycle
-
Consideration of key structural features
Medicinal chemists consider various structural aspects, including:
- Size and shape of the carbon skeleton: The overall bulk and spatial arrangement of the molecule.
- Functional groups: The presence and position of specific chemical groups that can participate in interactions like hydrogen bonding, ionic interactions, or hydrophobic interactions.
- Stereochemistry: The three-dimensional arrangement of atoms (e.g., enantiomers, diastereomers) can profoundly impact activity due to specific interactions with chiral biological targets.
- Substitutions: The nature and position of substituents on a parent scaffold.
- Physicochemical properties: Changes in properties like solubility, lipophilicity, pKa (acidity/basicity), polarity, and molecular flexibility.
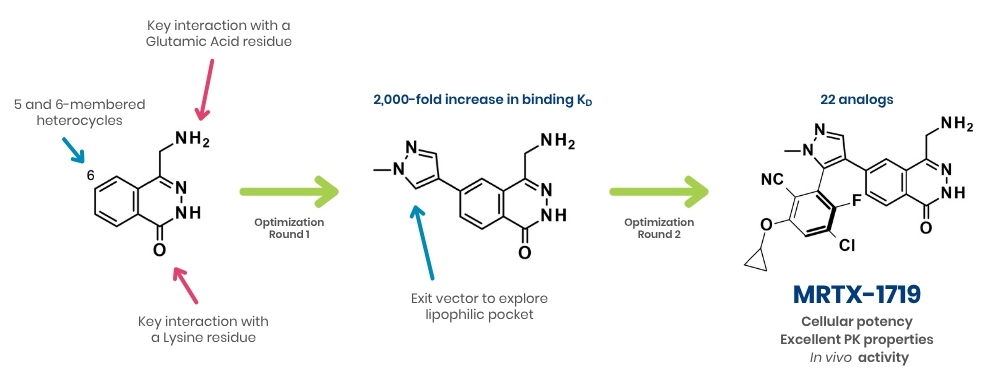
Pictured above: SAR studies of PRMT5-MTA inhibitor using a fragment-based approach.
-
Quantitative structure-activity relationship (QSAR)
QSAR is a more advanced approach that uses mathematical models to quantitatively relate specific physicochemical properties of a compound to its biological activity. This involves:
- Descriptor calculation: Quantifying various structural and physicochemical properties of molecules (e.g., hydrophobicity, electronic properties, steric bulk).
- Statistical modeling: Using statistical methods (e.g., regression analysis, machine learning) to build equations that predict activity based on these descriptors. QSAR allows for more precise predictions and a deeper understanding of the factors governing activity.
Structure-activity (SAR) analysis provides a roadmap for chemists to navigate vast chemical space, allowing them to systematically modify molecules to achieve desired biological outcomes, particularly in the pursuit of new and improved therapeutic agents.
To learn more about how SAR studies can support and de-risk your critical drug discovery pathway, discuss your project brief with our team today:


