
May 2023
The role of boron and benzoxaboroles as reversible covalent binders to target the serine and threonine proteome
1. Introduction
Historically, the pharmaceutical industry has tried to avoid making a covalent bond between a small molecule and the target protein. The risk of non-selective covalent binding to proteins other than the desired target, or an uncontrolled immune response occurring, was considered too risky, and too unpredictable. However, the cards have been reshuffled, as a number of drugs with a covalent binding mode of action have recently undergone clinical trials, or even been approved. Prime examples include the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (AbbVie), and the epidermal growth factor receptor (EGFR) inhibitor osimertinib (AstraZeneca), with sales totaling US$8.43 billion and $4.33 billion respectively in 2020. 1,2
The success of this approach highlights its potential as a strategy for hitting targets that are either difficult to address, or which are thought to be undruggable. Examples include Amgen’s sotorasib, an inhibitor of mutant KRAS(G12C). This GTPase had previously proved resistant to decades of drug discovery efforts.
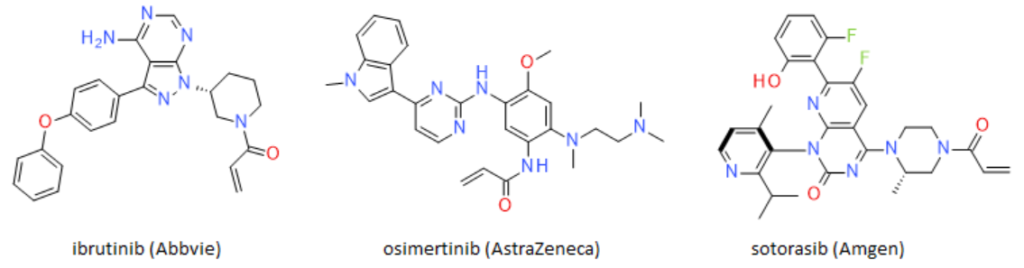
Figure 1: Structure of the covalent irreversible binders ibrutinib, osimertinib and sotorasib
It is worth taking a step back to consider the different mode of binding, and the potential advantages of covalent binders, whether irreversible or reversible, over non-covalent alternatives.
Protein modulators are molecules that bind to proteins, and prevent their normal activity. This is typically achieved either by blocking an enzyme’s active site directly, or by binding to a protein’s allosteric site, which causes a conformational change that prevents the protein from carrying out its function.
Different modes of binding
Protein inhibitors can interact with their targets via several different modalities:
- Competitive inhibition. Here, the inhibitor molecule binds to the active site of the enzyme, preventing the endogenous substrate from binding. This type of inhibition is in an equilibrium state, meaning that the inhibitor can be displaced if the concentration of the natural substrate is increased.
- Non-competitive inhibition. In this case, the inhibitor molecule binds to a site on the enzyme that is not the active site. Binding to this allosteric site causes a conformational change in the enzyme, preventing it from carrying out its function.
- Uncompetitive inhibition. The inhibitor molecule binds to the enzyme-substrate complex, preventing the substrate from being converted to its product.
- Mixed inhibition. Here, the inhibitor molecule can bind to both the enzyme and the enzyme-substrate complex. The resulting effects depend on the relative affinities of the inhibitor for the enzyme and the enzyme–substrate complex.
Covalent binding
Covalent irreversible inhibitors permanently bind to the target enzyme, leading to long-lasting inhibition, and shutting down the protein’s activity. This is particularly relevant when the target enzyme has a long half-life, as to regain activity, the inactivated enzyme must be replaced by new protein synthesised by the body.
In the case of competitive inhibition with an endogenous substrate, some enzymes have a high affinity for their substrate, and as a result, may be difficult to inhibit with reversible inhibitors at a physiologically acceptable concentration. Irreversible inhibitors can be more effective in these cases, as an irreversible binder will occupy the substrate’s binding site, even with a lower binding affinity, and concentration compared to the substrate, with similar selectivity.
- irreversible
Drugs that make covalent irreversible bonds with their target rely on reactions with specific amino acid residues in the protein, and a reactive electrophilic warhead. Generally, cysteine, histidine and lysine are the most likely to react with electrophilic compounds. Cysteine is particularly reactive, as it has a thiol (-SH) group that is readily oxidised and forms disulfide bonds, and reacts with electrophilic compounds via nucleophilic attack. Histidine and lysine are also relatively reactive because the nitrogen atoms in their side chains can act as nucleophiles in certain reactions. It is, however, important to consider the environment surrounding the amino acid being targeted, as its reactivity can be influenced by pH and the presence of ions or cofactors.
Several advantages can result if a covalent binder is used. Efficacy can be improved, lower doses could be required, patient compliance may be improved through less frequent dosing, and the emergence of resistance may be reduced. Many covalent inhibitors currently undergoing Phase 2 and Phase 3 clinical trials target cysteine residues using an acrylamide warhead to bind to the target protein, as illustrated in Figure 1. However, this may limit the covalent proteome space, as cysteine is not commonly found in binding sites, and is mainly accessible in intracellular proteins. In addition, potential toxicities mediated by unpredictable modification of off-target cysteines represents a possible hurdle for expansion of covalent irreversible drug programs.
Can we move beyond cysteine, and do we have the tools in the toolbox that we will need for this?
- reversible
Covalent reversible binders (CRBs) are a class of compounds that have gained attention for targeting hydrolase enzymes with a catalytic serine, threonine, or cysteine residue. A reversible covalent strategy has the potential to mitigate the risk of toxicities arising from non-specific irreversible binding or haptenization, while still maintaining the selectivity benefits associated with addressing a specific nucleophilic amino acid. It also allows the on-target residence time to be fine-tuned.
CRBs form a covalent bond with the target protein. This bond is reversible, meaning it can be broken and reformed, while still enabling tight binding of the compound to the target protein. In the field of kinase drug discovery, Serafimova et al.3 have demonstrated the use of “hyperactivated” Michael acceptors, such as the α-cyanoacrylamides, where the increased α–CH acidity of the cysteine adduct promotes reversibility.
2. Boron based drugs
History
Boron, and benzoxaboroles in particular, have significant potential as CRBs. Boron-based drugs have been studied for a variety of diseases, including cancer, autoimmune conditions and infectious diseases (Scheme 1).
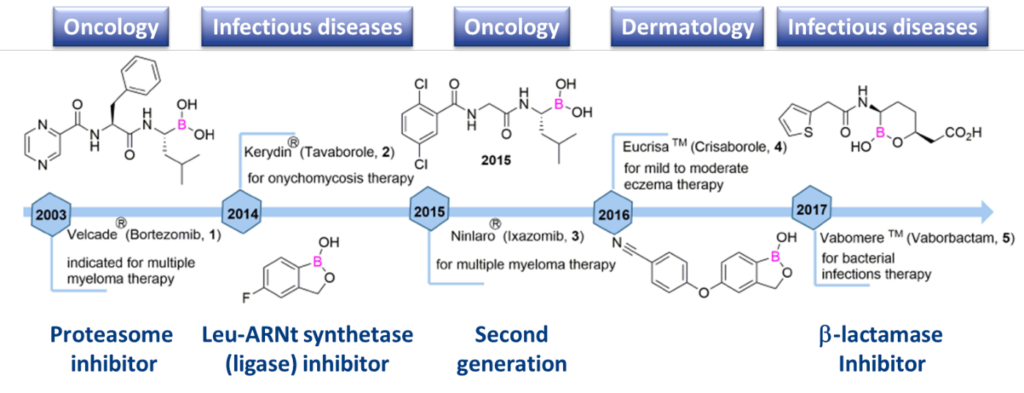
Specificity
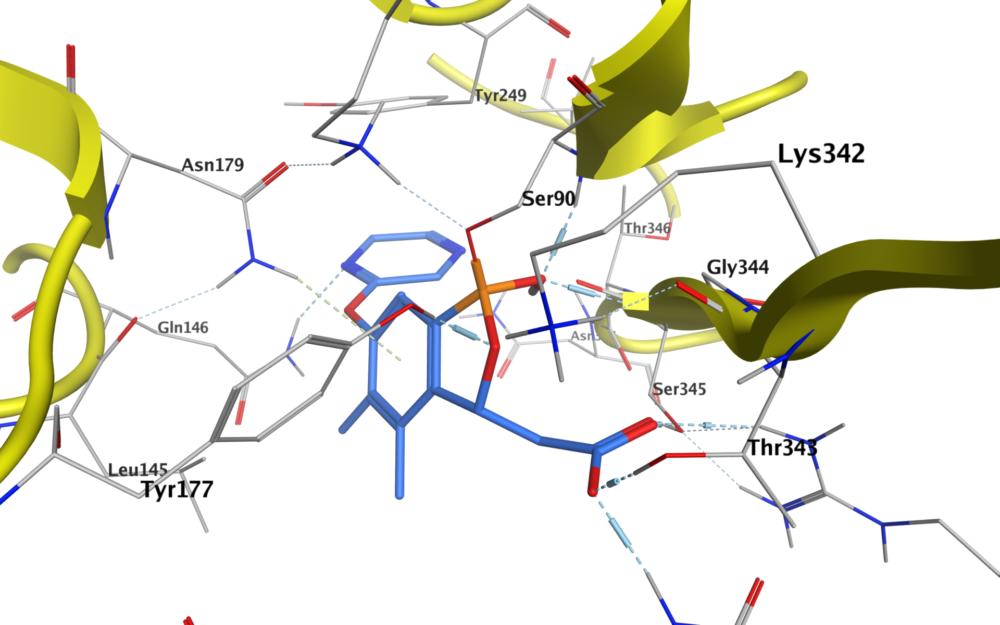
A specific feature of boron that makes it an attractive alternative as a covalent reversible warhead is that its compounds readily interconvert between neutral trigonal planar sp2 and tetrahedral sp3 hybridization states. This allows the compounds to adopt a diverse range of binding modes during target engagement, forming covalent adducts with nucleophiles such as serine, lysine, tyrosine, threonine and cysteine residues in target proteins. Benzoxaboroles (also known as oxaboroles) are of particular interest, as the boronic ester is a better Lewis acid than boronic acid. The enhanced Lewis acidity is likely a result of ring strain in the five-membered ring that gives an sp3 hybridized atom upon serine binding.5
The stabilized complex that is formed can then engage in additional hydrogen bonding with adjacent lysine or tyrosine residues. In addition, benzoxaboroles are attractive molecules for medicinal chemists, as the ester rings are generally more hydrolytically stable than boronic acids, and often have good solubility and reasonable permeability. In addition, good stability to metabolism by microsomes or the S9 liver fraction from a range of species has been observed.
3. Oncodesign Services library
The advantages and attractiveness of this scaffold led us to design and synthesize, an initial library set of 272 benzoxaboroles, including fragments. This was possible because of the previous synthetic experience of our chemists.
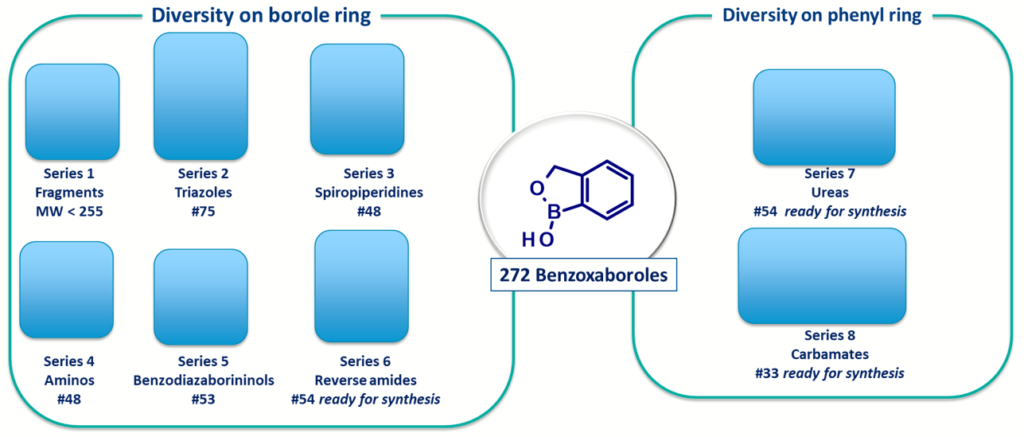
Scheme 2: Library of benzoxaborole scaffolds available for screening at Oncodesign Services
Careful attention was taken when designing the library to maintain low molecular weight and logD. We also ensured they had attractive hit-like properties for initial hit finding campaigns (see Scheme 3).
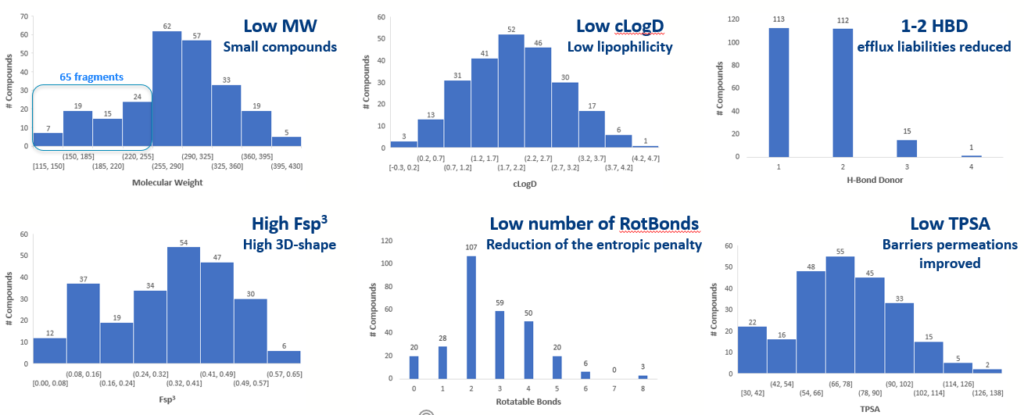
Scheme 3: Phys Chem and Ro5 properties of the Benzoxaboroles scaffold library
The success of this initial exercise led us to extend the library, adding additional series. We now have 477 designed compounds ready to be synthesized, complementing the chemical space of the initial set. (See Scheme 4). Using all these different series, a virtual extension has been designed, based on a selection of Enamine building blocks. This led to an SDF file of 6990 compounds that are now available for virtual screening.
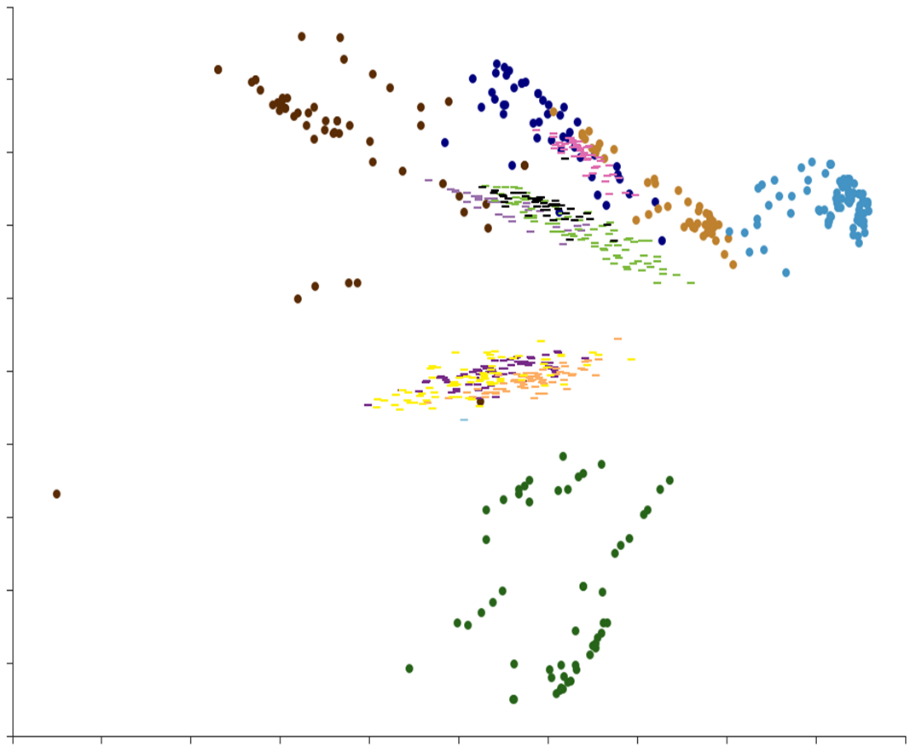
Scheme 4: Chemical space of the library of benzoxaboroles at Oncodesign Services: 272 synthesized compounds (●) and 477 compounds to be synthesized (-)
When comparing the newly created chemical space of the benzoxaboroles library with the chemical space from the library of commercially available covalent binders, it was clearly complementary to the space occupied by cysteine targeted warheads.
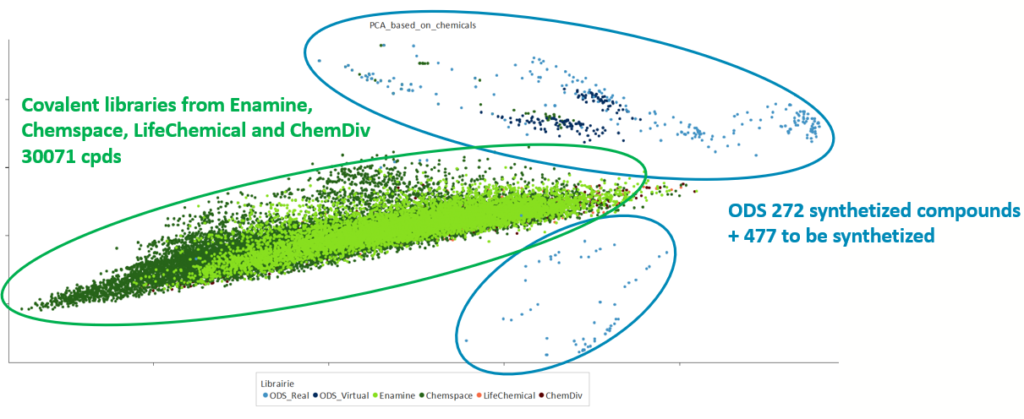
To go further, conclusion
Overall, boron has shown promise as a reversible covalent binder in drug discovery. Its ability to selectively target specific proteins or enzymes in the body, and its ability to create drugs that are highly selective and specific, make it a useful tool for developing new drugs to treat a variety of diseases.
Despite the potential benefits of using boron as a reversible covalent binder in drug discovery, there are still challenges to be overcome, and the development of boron-based drugs certainly requires practical synthetic knowledge. Additional research is needed to fully explore the benefits associated with the use of boron in drug discovery. The ability to form covalent bonds with oxygen – especially serine6 and threonine, but also RNA – present opportunities for potency that exceeds what can be achieved without covalency.
If you are interested in challenging your target with an initial set of benzoxaboroles, please get in touch. All compounds are at 10 mM in DMSO solution and 96-well plates, ready to be screened.
About the author
| This article was written by Christophe Parsy PhD, MBA, head of medicinal chemistry at Oncodesign Services. Christophe has more than 20 years of drug discovery experience. He has held several positions throughout his career, including Biofocus, Idenix Pharmaceuticals, MSD and Deinove, and joined Oncodesign Services in 2019. Christophe holds a PhD in organic chemistry from the University of Liverpool, and an executive MBA from the Montpellier Business School. |  |
-
References
1- Lonsdale, R. & Ward, R. A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 47, 3816–3830 (2018).
2- Sagonowsky, E. The top 20 drugs by worldwide sales in 2020. Fierce Pharma https://www.fiercepharma.com/special-report/top-20-drugs-by-2020-sales (2021).
3- Serafimova et al. Nat Chem Biol. 2012 Apr 1;8(5):471-6.
4- Song et Al., Acta Pharmaceutica Sinica B, Volume 11, Issue 10, October 2021, Pages 3035-3059
5- Leach, A. Annual Reports in Medicinal Chemistry, Volume 56, 2017, chapter five, Boron and covalent inhibition
6- Martin, J.S., et al., Bioorganic & Medicinal Chemistry 27 (2019) 2066–2074
Do you want to discuss with our expert ? Contact him!